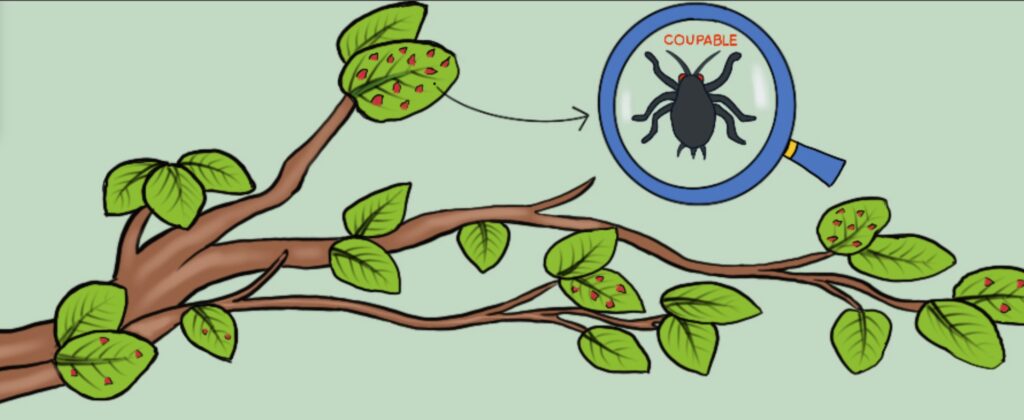
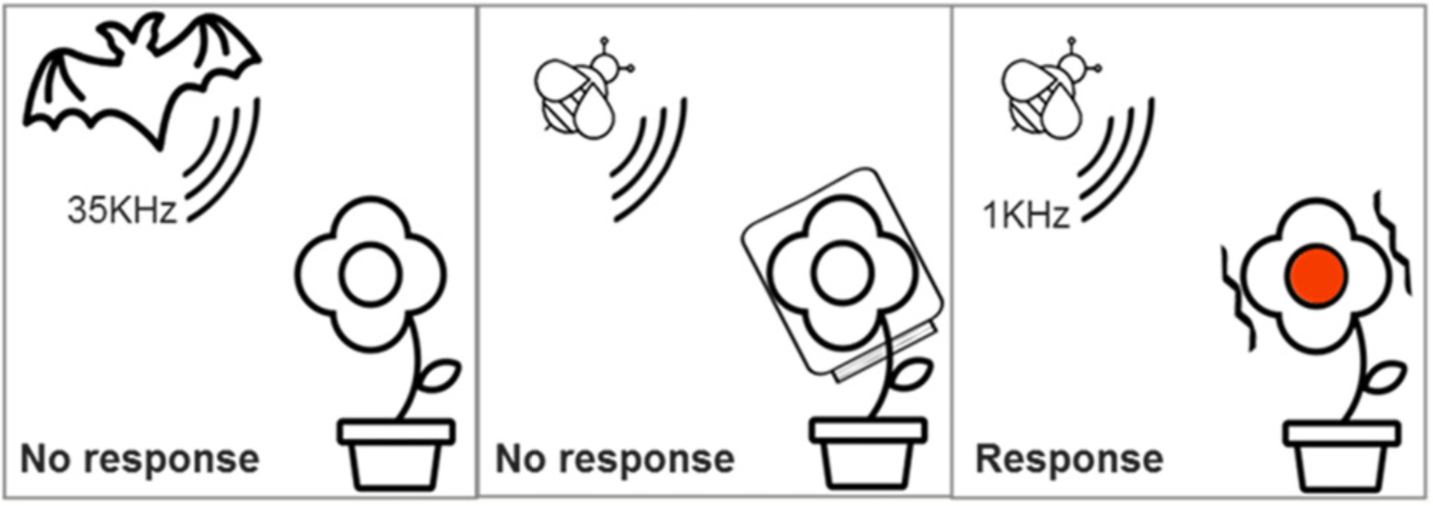
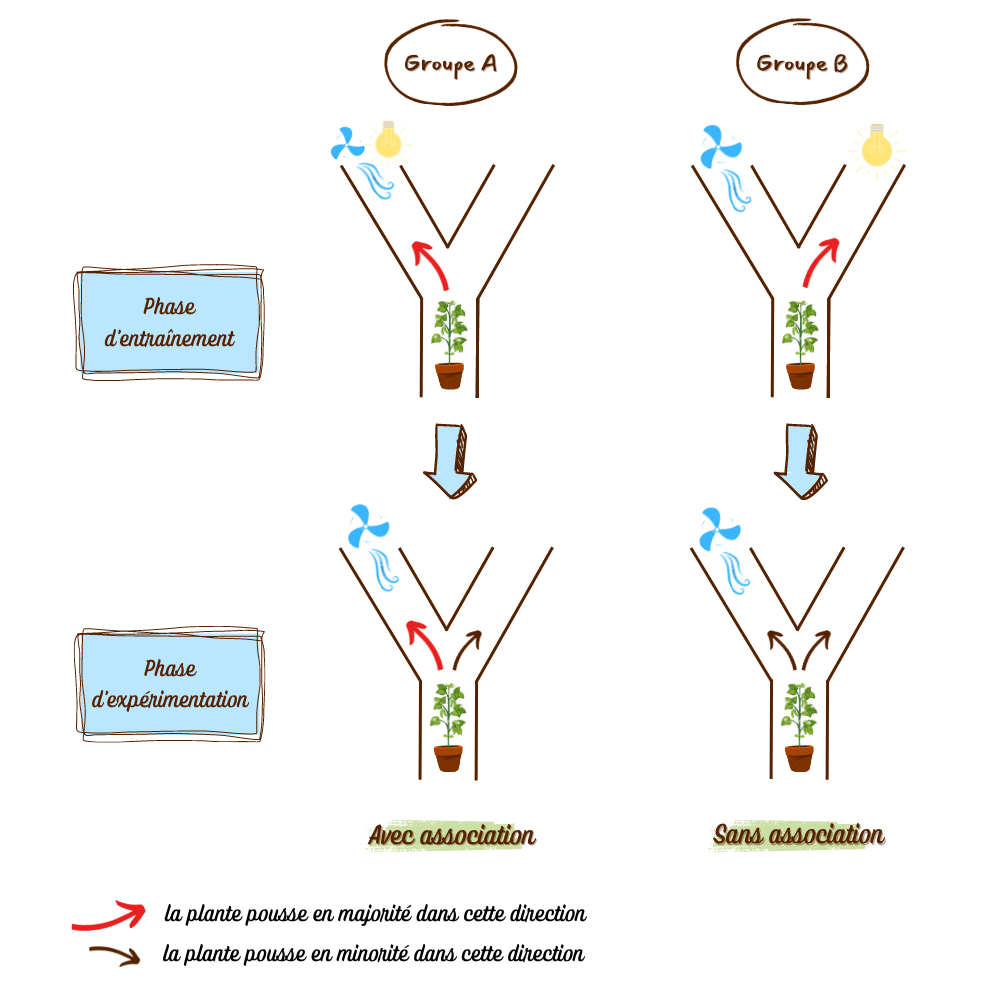
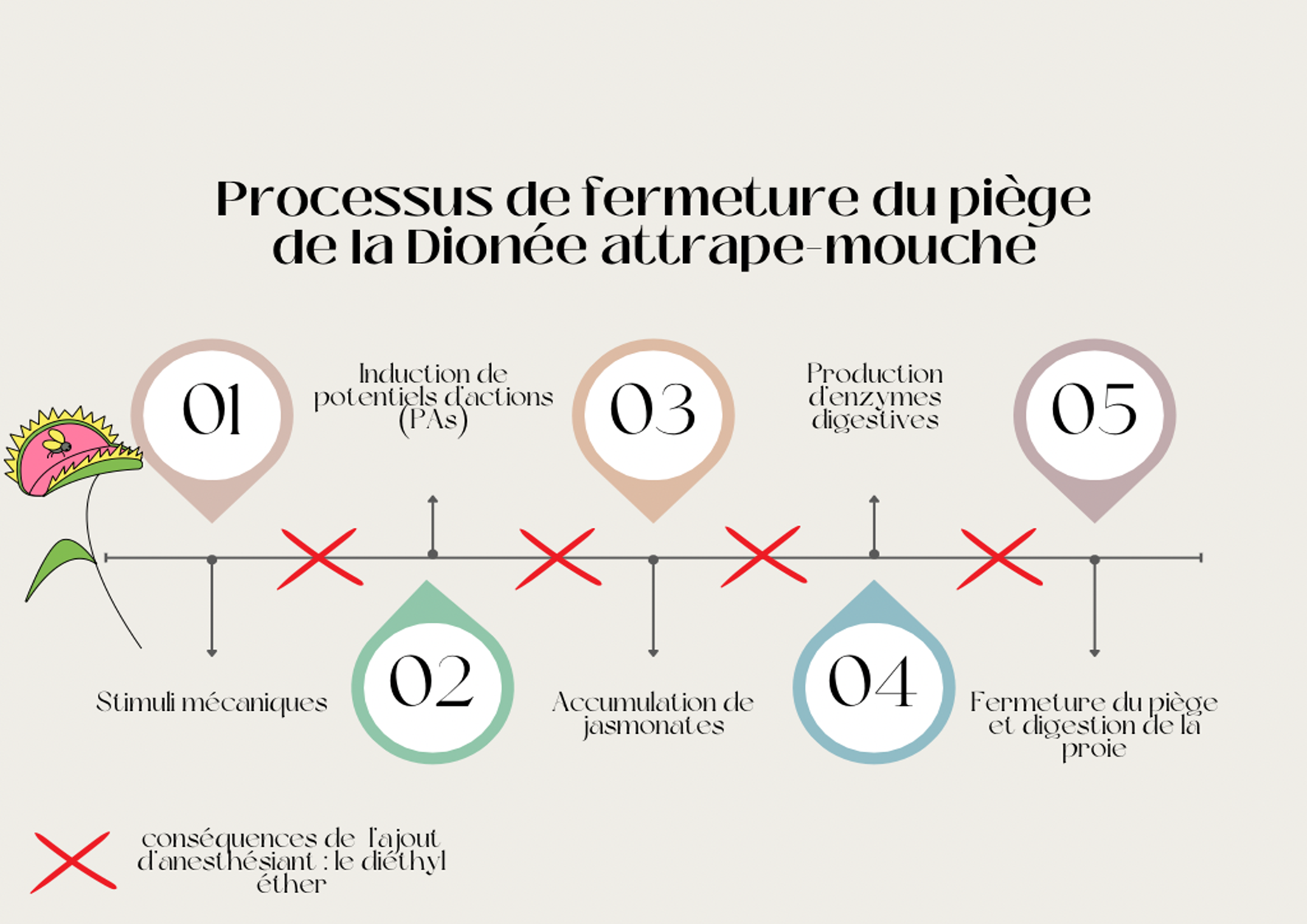
La galle et le puceron: une fable d’actions Chez les végétaux, une galle est une excroissance d’un de ses tissus, produite dans certains cas sous l’action d’un... Lire la suite Pourquoi les plantes ont-elles de l’acné ?- CopiePourquoi les plantes ont-elles de l’acné ?| MEG La galle et le puceron: une fable d’actions- CopieLa galle et le puceron: une fable d’actions| Bionum Les plantes, sourdes comme des pots ?- CopieLes plantes, sourdes comme des pots ?| Bionum Idotées, les abeilles de l’océan ? - CopieIdotées, les abeilles de l’océan ? | Bionum L’apprentissage par association chez les plantes en quête de lumière- CopieL’apprentissage par association chez les plantes en quête de lumière| Bionum Dionée qui dort… ne dîne pas- CopieDionée qui dort… ne dîne pas| Bionum La dionée dans les bras de Morphée ?- CopieLa dionée dans les bras de Morphée ?| Bionum Breaking news : des scientifiques s’amusent à endormir des champignons !- CopieBreaking news : des scientifiques s’amusent à endormir des champignons !| Bionum OSEF : Objets Scientifiques Etonnants et Fascinants- CopieOSEF : Objets Scientifiques Etonnants et Fascinants| Bionum Tous les billets de blog Agenda Restitution CBioNum 202431 mai 2024Restitution CBioNum 2024 Tout l’agenda Partenaires